8.4: Evaluation of new fresh frozen plasma/cryoprecipitate components for transfusion
8.4.1: Introduction
In establishing any novel component, the development process is expected to involve three stages (see Table 8.1d):
- Investigation (Phase 0): Initial intensive investigation of a range of parameters on a relatively small number of units (e.g. 10 -16) to establish concepts. This should involve in vitro studies with serial sampling, and may also involve in vivo studies. Components produced during this phase should not be used for transfusion. For clarity the guidance on which tests need to be performed is as shown in Table 8.4.
- Validation (Phase 1/Phase 2): Operational validation on a larger number of units (e.g. 125) to establish routine operation of the technique, normally testing for those parameters listed in the current edition of the Red Book. These tests may be supplemented by a limited set of assays selected from the investigational phase to allow setting of routine quality parameters. This may involve in vivo studies and normally would involve sampling at the times shown below for routine testing.
- Routine (Local process validation): Ongoing routine validation using a small set of parameters selected on the basis of the above studies. This will not normally involve in vivo studies. Advice may be sought from SACBC on the validation requirements for plasma components produced from automated processing of whole blood or other technologies that are not specified in Table 8.4.
8.4.2: In vitro evaluation of novel fresh frozen plasma
8.4.2.1: Suggested study design
Because of the wide normal range of some clotting factors and potential inter-batch variation of assays, it is suggested that novel units and controls be produced and assayed in parallel, with the novel technology being the only variable. A less costly alternative, if logistics permit, is to do a pooled paired comparison, where two units are pooled, and one half processed by the novel technique. This provides greater statistical power for fewer units assayed, and is particularly important for storage studies. Since levels of FVIII and von Willebrand factor are ABO group-dependent, investigators should consider an equal mix of group A and O donations in the experimental design. The number of units to be studied should be based on the study objectives and design, and determined by statistical analysis based on the difference between test and control units to be detected. A sample size of at least 16 test or controls would be required to detect a 30% difference in FVIII levels using an unpaired study. Fewer units will be required if a pooled and split study design is used.
For leucocyte depleted or pathogen reduction systems it is recommended that assays are performed on samples collected before and after the process under investigation. Ideally provision should be made for storing and testing aliquots from each pack at every time point, as thawing out three or four different packs at each time point introduces excessive variation. However, a pre-validation should be done to ensure that the behaviour of the aliquoted component during storage is the same as that in the main pack.
8.4.2.2: Assays required
The extent of any evaluation depends in part on the degree of novelty of the component. The list of assays below need not be applied in every setting. Table 8.4 gives a summary of which assays are recommended in different situations. Advice may be sought from SACBC on the validation requirements for plasma components produced from automated processing of whole blood or other technology that is not specified in Table 8.4.
All evaluations must include the routine quality control parameters such as FVIII:C.
Before freezing:
- volume, platelet count, WBC*, RBC, total protein
- prothrombin time (PT), activated partial thromboplastin time (APTT)
- factors I (fibrinogen), II, V, VII, VIII, IX, X, XI, XIII, von Willebrand factor (vWf):Ag, vWf:RiCof, which measures the functional activity or an assay validated as yielding equivalent results, vWf multimeric analysis, vWF cleaving protease
- inhibitors of coagulation – antithrombin, protein C, protein S, α2-antiplasmin
- markers of unwanted activation of coagulation* – prothrombin fragment 1.2, fibrinopeptide A, factor XIIa, thrombin-antithrombin (TAT) complexes.
*Particularly relevant to plasma which has been collected by any filtration technique, in which case the assays should be performed before and after filtration or to packs made of novel materials.
During storage:
- Consideration should be given to performing storage studies at ≥20°C in addition to those at ≤30°C to reflect hospital storage conditions. Samples should be taken at 12 and 24 months. Ideally, all clotting factors should be assayed at each time point, if only in a few packs. FVIII should be assayed at each time point as a minimum in addition to the proteins most severely affected by the initial process.
- Storage parameters may be assayed after the date of implementation of routine production, provided data ‘keep ahead’ of the age of any clinical product which might be issued.
Novel Plasticisers (please also refer to Table 8.4 and section 8.8 for further guidance):
Where novel plasticisers are used, the levels of recovered plasticiser should be monitored over shelf life to assess the levels of leaching into the blood component. Blood bag manufacturers or external laboratories may be required for chemical analysis of plasticisers. Methodology will be specific to the plasticiser under investigation, but likely to be by liquid chromatography-mass spectrometry. Advice can be sought from manufacturers, SACBC and peer-reviewed literature. It is also important to consider metabolites that may also influence product quality and may have toxicological effects. Concentrations in the plasma should be measured at the beginning, during and end of storage to assess leaching and potential patient exposure. Consideration must be given to the effects of irradiation on the bag and subsequent leaching potential. Suppliers must undertake toxicology studies as part of CE/UKCA/UKNI marking. Suppliers must provide evidence of an independent review of toxicology data; this data will then be reviewed by SACBC.
8.4.3: In vitro evaluation of novel cryoprecipitate
It is assumed that this will be produced from a ‘novel’ start plasma so that investigators will be aware of any specific losses of clotting factors which should be particularly considered.
Assays to be performed before and after production, and during storage: fibrinogen, FVIII:C.
8.4.4: Cryosupernatant
The only clinical indication for this component is for plasma exchange procedures for patients with thrombotic thrombocytopenic purpura. Analysis of vWf multimers and cleaving protease is therefore appropriate. vWf multimeric and cleaving protease analysis should be performed in a laboratory recognised to be proficient in this technique and which is performing the assay regularly.
Table 8.4 Evaluation of novel plasma components
| Fresh frozen plasma | Cryo-precipitate | Cryo-supernatant | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameter | Novel filter | New centrifuge/ component extractor | Novel anticoagulant | Novel plasticiser /plastic | Novel apheresis system | Novel apheresis + anticoagulant | Pathogen reduction | ||
| Volume | 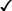 |
|
|
|
|
|
|
|
|
| Leucocyte content | |
|
|
|
|
|
– | ||
| FVIII:C | |
|
|
|
|
|
|
|
– |
| Platelets | |
|
|
|
|
|
|
– | – |
| PT ratio | |
– | |
|
– | |
|
– | – |
| ROTEM/ROTEG | |
– | |
|
– | |
|
– | – |
|
APTT ratio |
|
– |
|
|
– |
|
|
– |
– |
|
Fibrinogen |
|
– |
|
|
– |
|
|
|
– |
|
II, V, VII, IX, X, XI, XIII |
|
– |
|
|
– |
|
|
– |
– |
|
vWf:Ag |
|
– |
|
|
– |
|
|
|
– |
|
vWf:RiCof |
|
– |
|
|
– |
|
|
|
|
|
AT III, Prot C, Prot S |
|
– |
|
|
– |
|
|
– |
– |
|
TAT/Frag1.2/FPA + FXIIa |
|
– |
|
|
|
|
|
Omit if not elevated in source plasma |
|
|
C1 inhibitor |
|
– |
|
|
– |
|
|
|
– |
|
vWf multimers |
|
– |
|
|
– |
|
|
|
|
|
vWF cleaving protease |
|
– |
|
|
– |
|
|
|
|
|
Alpha-2 anti-plasmin |
|
– |
|
|
– |
|
|
|
– |
|
Pathogen reduction* |
|
|
|
|
|
|
|
|
– |
|
PrPc /microvesicles |
? |
– |
|
|
|
|
|
|
– |
|
Clinical trial |
– |
– |
# |
|
# |
# |
|
# |
# |
| Recovered plasticiser in supernatant | |
||||||||
|
Key: |
|||||||||
8.4.5: In vivo studies
Whether or not in vivo studies are needed depends on the degree of novelty of the component, e.g. this may not be necessary for plasma which has been leucocyte depleted in the course of producing leucocyte-depleted red cells, but would certainly apply in the case of a novel pathogen reduced plasma which had been exposed to chemicals. Unlike red cells and platelets, administration to normal volunteers has not been traditional. Suitable patient groups to consider would be:
For fresh frozen plasma:
- correction of prolonged international normalised ratio (INR) prior to liver biopsy
- liver transplant recipients
- plasma exchange for thrombotic thrombocytopenic purpura (TTP)
- disseminated intravascular coagulation (DIC).
It is difficult to get permission to study neonates and usually considerable experience has to have been gained with the product in adults.
A randomised design is preferred, with standard fresh frozen plasma as control.
For cryoprecipitate:
- DIC
- liver disease/transplant
- congenital hypofibrinogenaemia, if maintained on cryoprecipitate.